

Protein Language Models (PLMs) have emerged as powerful tools in bioinformatics, trained on vast protein databases to predict amino acid sequences and generate functional representations of proteins. These models excel at tasks such as predicting protein folding and mutation outcomes, largely due to their ability to capture conserved sequence motifs, which are critical for protein health. However, the relationship between sequence conservation and protein function can be influenced by evolutionary and environmental factors, adding complexity to the model’s predictions. To overcome these challenges, PLMs typically rely on pseudo-likelihood objectives. Incorporating additional data sources, such as textual annotations detailing protein functions and structures, has been identified as a promising strategy to enhance their performance.
The PAIR Framework: Integrating Textual Annotations for Improved Protein Prediction
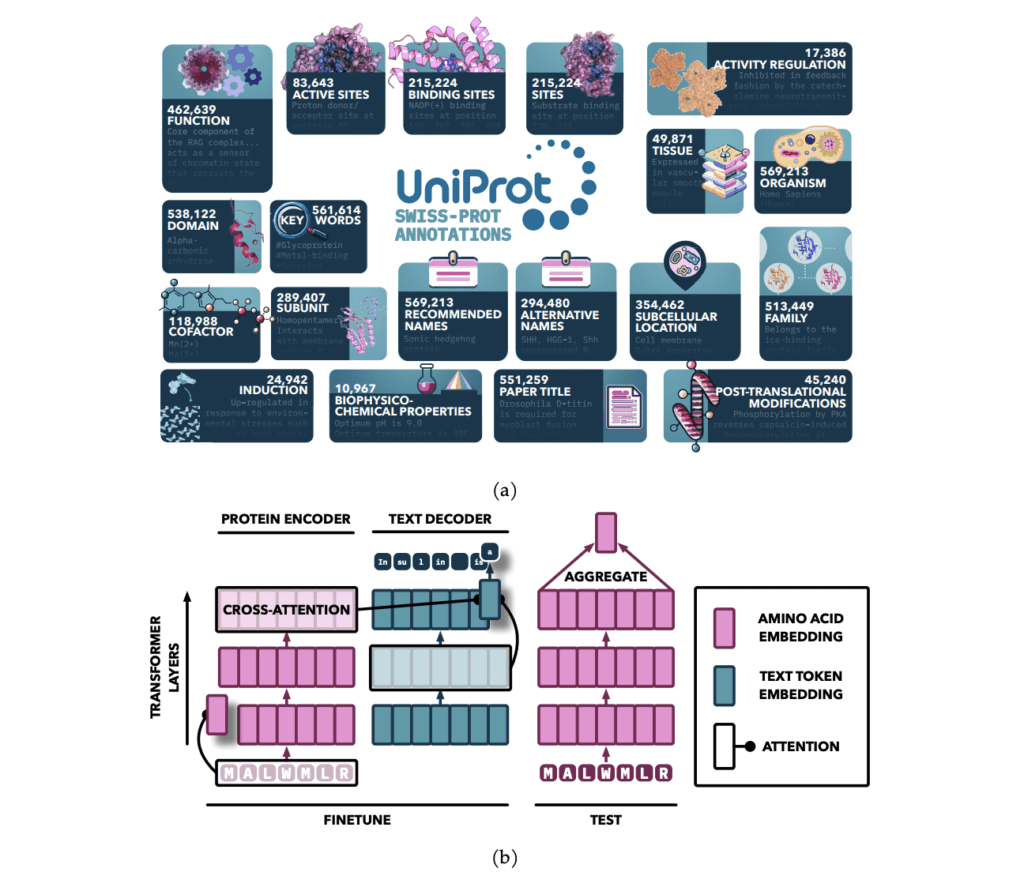
A team from the University of Toronto and the Vector Institute developed a framework to enhance PLMs by integrating textual annotations from UniProt, focusing on 19 types of expert-curated data. This new approach, named Protein Annotation-Improved Representations (PAIR), uses a text-based decoder to guide the model’s learning process. By fine-tuning PLMs with these textual annotations, PAIR significantly improves the models’ performance, especially in tasks related to protein function prediction. Notably, PAIR outperforms traditional methods like the BLAST search algorithm, particularly for proteins that show low sequence similarity to the training data.
The integration of textual annotations into PLMs marks a departure from the traditional sequence alignment-based methods like BLAST, which rely on detecting protein homology through sequence similarities. These classical methods work well for sequences that are highly similar but struggle with distant homology detection. PLMs, on the other hand, apply deep learning techniques to learn protein representations from large-scale sequence data, akin to natural language processing models. The PAIR framework builds on these advancements, offering a more accurate and robust method for protein function prediction.
How PAIR Works: Sequence-to-Sequence Model with Cross-Attention
The PAIR framework employs an attention-based sequence-to-sequence architecture. The encoder processes protein sequences into fixed-length representations using self-attention, a technique that allows the model to weigh the importance of different parts of the sequence. The decoder then generates textual annotations in an auto-regressive manner, which are used to predict protein functions. Pretrained models, such as ProtT5 and ESM, serve as the encoder, while SciBERT is used as the text-based decoder.
The model is trained on a range of annotation types from databases like Swiss-Prot, utilizing a specialized sampling technique for robust training. The training process is carried out on high-performance computing (HPC) clusters with multi-node setups, using bfloat16 precision for efficiency. This allows the model to scale effectively while ensuring accurate predictions.
Results and Benefits of the PAIR Framework
PAIR has demonstrated superior performance over traditional methods like BLAST, particularly for proteins with low sequence similarity to the training data. The inclusion of textual annotations, such as those found in Swiss-Prot and UniProt, provides a rich source of additional information that helps the model better understand protein functions. The integration of cross-attention between the encoder and decoder further improves the relationship between the protein sequences and their corresponding annotations.
By fine-tuning PLMs with these annotations, PAIR is able to predict a wide variety of functional properties for proteins, including those that were previously uncharacterized. This capability is particularly valuable for identifying functions in proteins that have low homology to known sequences. PAIR’s strong generalization ability means that it can be applied to novel tasks and datasets, making it a powerful tool for bioinformaticians.
Future Directions and Potential Applications
While PAIR has already shown substantial improvements in protein function prediction, it opens the door for further advancements in protein representation learning. The ability to incorporate other types of data, such as 3D structural information or genomic data, could provide even richer representations of proteins, enhancing the model’s predictive capabilities.
Moreover, the PAIR framework’s design is flexible and could be applied to other biological entities, such as small molecules or nucleic acids, expanding its potential uses in various bioinformatics applications.
Conclusion
The PAIR framework is a significant step forward in improving protein language models by integrating textual annotations alongside traditional sequence-based data. Its ability to predict protein functions more accurately, especially for low-homology sequences, represents a major advancement in the field. PAIR demonstrates the power of combining different data modalities to create more robust and reliable models for protein function prediction, and its flexibility offers promise for broader applications in bioinformatics. As the field continues to evolve, PAIR could play a crucial role in advancing our understanding of proteins and their complex functions.
Last Update: November 24, 2024



