

Protein language fashions (PLMs) are educated on huge protein databases to predict amino acid sequences and generate perform vectors representing proteins. These fashions have confirmed useful in quite a few functions, akin to predicting protein folding and mutation outcomes. A key motive for his or her success is their functionality to grab conserved sequence motifs, that are typically important for protein well being. Nonetheless, evolutionary and environmental elements can have an effect on the connection between sequence conservation and well being, making it difficult. PLMs depend upon pseudo-likelihood objectives, nevertheless incorporating additional info sources, akin to textual content material annotations describing protein options and buildings, may improve their accuracy.
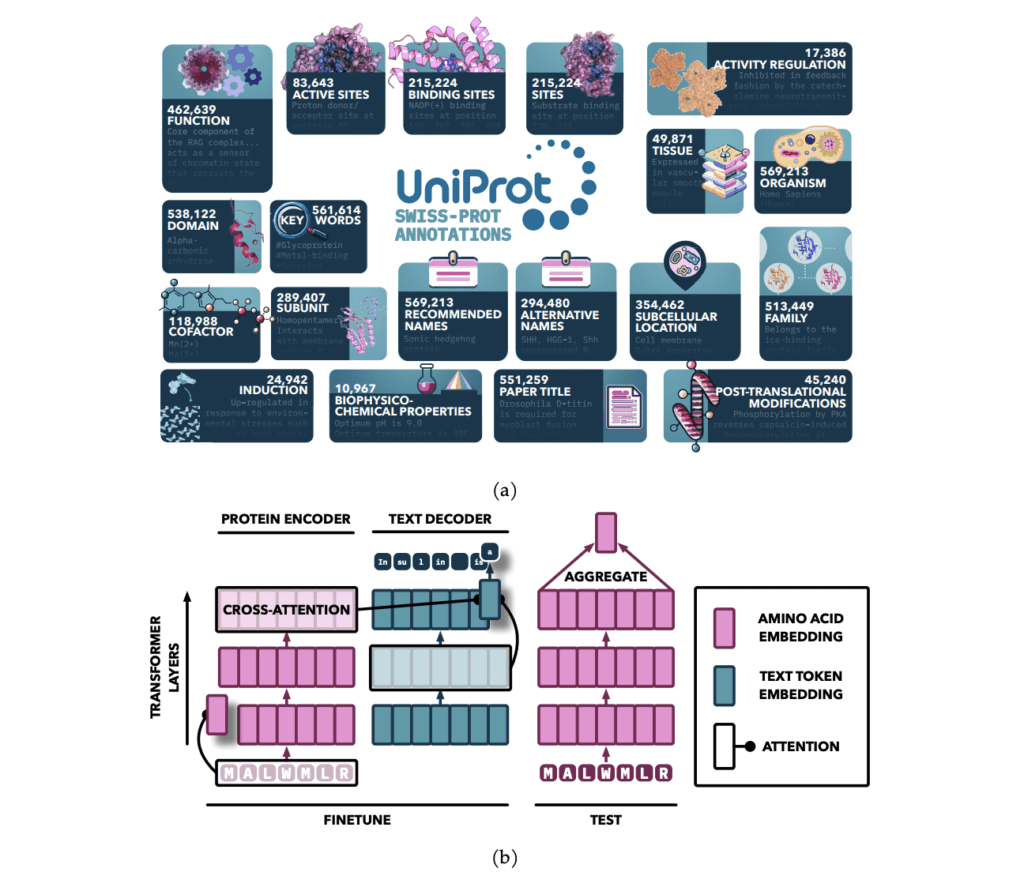
Researchers from the Faculty of Toronto and the Vector Institute carried out a analysis that enhanced PLMs by fine-tuning them with textual content material annotations from UniProt, specializing in nineteen kinds of expert-curated info. They launched the Protein Annotation-Improved Representations (PAIR) framework, which makes use of a textual content material decoder to info the model’s teaching. PAIR significantly improved the fashions’ effectivity on function prediction duties, even outperforming the BLAST search algorithm, notably for proteins with low sequence similarity to teaching info. This technique highlights the potential of incorporating varied text-based annotations to advance protein illustration finding out.
The sector of protein labeling traditionally will depend on methods like BLAST, which detects protein sequence homology by way of sequence alignment, and Hidden Markov Fashions (HMMs) that incorporate additional info akin to protein family and evolutionary data. These classical approaches perform properly with sequences of extreme similarity nevertheless wrestle with distant homology detection. This drawback has led to the occasion of PLMs, which apply deep finding out methods to be taught protein representations from large-scale sequence info impressed by pure language processing fashions. Newest developments moreover mix textual content material annotations, with fashions like ProtST leveraging varied info sources to reinforce protein function prediction.
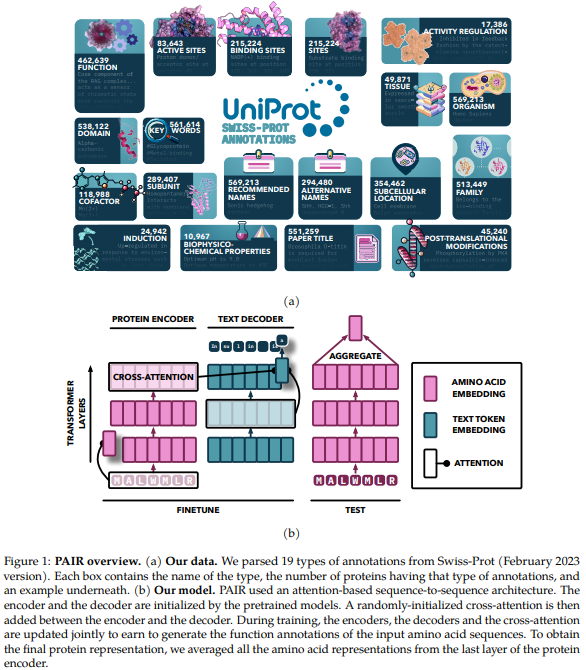
The model makes use of an attention-based sequence-to-sequence construction, initialized with pretrained fashions and enhanced by together with cross-attention between the encoder and decoder. The encoder processes protein sequences into regular representations using self-attention, whereas the decoder generates textual content material annotations in an auto-regressive technique. Pretrained protein fashions from the ProtT5 and ESM households perform the encoder, whereas SciBERT is the textual content material decoder. The model is educated on plenty of annotation kinds using a specialised sampling technique, with teaching carried out on an HPC cluster using multi-node teaching with bfloat16 precision.
The PAIR framework enhances protein function prediction by fine-tuning pre-trained transformer fashions, like ESM and ProtT5, on high-quality annotations from databases like Swiss-Prot. By integrating a cross-attention module, PAIR permits textual content material tokens to maintain amino acid sequences, bettering the connection between protein sequences and their annotations. PAIR significantly outperforms typical methods like BLAST, notably for proteins with low sequence similarity, and reveals sturdy generalization to new duties. Its functionality to take care of restricted info conditions makes it a treasured instrument in bioinformatics and protein function prediction.
The PAIR framework enhances protein representations by utilizing varied textual content material annotations that seize essential purposeful properties. By combining these annotations, PAIR significantly improves the prediction of varied purposeful properties, along with these of beforehand uncharacterized proteins. PAIR consistently outperforms base protein language fashions and traditional methods like BLAST, notably for sequences with low similarity to teaching info. The outcomes advocate incorporating additional info modalities, akin to 3D structural data or genomic info, may enrich protein representations. PAIR’s versatile design moreover has potential functions for representing completely different natural entities, akin to small molecules and nucleic acids.



